
I database delle industrie farmaceutiche e dei promotori europei contengono notevoli quantità di dati clinici raccolti durante le sperimentazioni cliniche in tutto il mondo. Questi dati in Europa sono oggi bloccati dal GDPR e da norme stringenti che non permettono la restituzione dei dati ai pazienti stessi e al loro riuso per ulteriori attività di ricerca. Questo limite comporta inevitabilmente un limitato accesso ai propri dati clinici da parte dei pazienti, che non vengono quindi a conoscenza del loro quadro clinico. Il superamento di questo limite, attraverso la definizione di nuovi standard europei, è lo scopo di alcune iniziative progettuali.
Autore: Dott. Stefano Brumat, Università di Bologna (pagina personale)
Durante gli studi clinici (interventistici e non) viene raccolta una grande quantità di dati sanitari di alta qualità, ma – al di là degli obiettivi immediati dello studio – questi preziosi dati non vengono utilizzati nella misura che meritano. I dati raccolti, infatti, non possono essere utilizzati al di fuori dell’ambito della sperimentazione stessa. Attualmente (giugno 2022) sono registrati 27.049 studi clinici completi e 16.029 studi attivi in Europa (dati dal portale EU Clinical Trial Register ).
LE REGOLE/NORMATIVE VIGENTI
I dati di queste migliaia di studi potrebbero essere utilizzati per arricchire le cartelle cliniche dei pazienti, al fine di migliorare il processo decisionale clinico e ridurre la duplicazione delle procedure/indagini a carico dei pazienti e del sistema sanitario. Inoltre, la restituzione dei dati ai pazienti raccolti durante gli studi clinici potrebbe consentire loro di contribuire, con i propri dati, a ulteriori ricerche scientifiche, in particolare per le malattie rare in cui i trattamenti e i riferimenti clinici sono scarsi o non disponibili. Al contempo, vi è una crescente consapevolezza della necessità di una maggiore trasparenza da parte dei detentori dei dati (titolari del trattamento, ai sensi del GDPR), di un maggiore coinvolgimento dei partecipanti agli studi nella ricerca clinica e anche dell’importanza della restituzione dei dati delle sperimentazioni cliniche ai partecipanti agli studi.
Diversi sono gli ostacoli alla restituzione dei dati delle sperimentazioni cliniche ai partecipanti allo studio dovuti da limiti del sistema tecnico e normativo:
- la complessità nel determinare gli standard dei formati di catalogazione dei dati, i flussi tra i vari attori e la presenza di infrastrutture dedicate alla gestione di tali dati (database);
- la difficoltà a definire standard di condivisione dei dati tra gli Stati Membri;
- la complessità delle regole di gestione delle sperimentazioni cliniche (Consiglio Internazionale per l’Armonizzazione dei Requisiti Tecnici dei Farmaci per Uso Umano – ICH),in combinazione con le regole sulla Buona Pratica Clinica (GCP) e del Regolamento sulla Sperimentazione Clinica dell’Unione Europea (CTR);
- la mancanza di armonizzazione sia del quadro giuridico relativo al trattamento dei dati sanitari nei vari Stati Membri e la necessità di ulteriori orientamenti su alcuni aspetti pratici dell’attuazione del Regolamento Generale sulla Protezione dei Dati (GDPR), sia per l’uso primario e/o secondario dei dati personali raccolti durante la sperimentazione clinica.
Senza dimenticare l’influenza dei dubbi e delle limitazioni al consenso da parte dei pazienti, a causa del timore alla mancanza di trasparenza da parte di chi gestisce i propri dati.
Per superare questa empasse è necessario definire nuovi standard, a livello europeo, per la restituzione dei dati ai singoli partecipanti allo studio e per definire l’utilizzo secondario dei dati clinici ai fini della ricerca clinica. Processo che deve tenere conto di svariati aspetti:
- la conformità alla normativa europea e nazionale sulla tutela dei dati personali;
- l’opinione dei partecipanti allo studio, delle organizzazioni dei pazienti, degli sperimentatori e degli operatori sanitari;
- le opinioni delle diverse autorità di regolamentazione (garanti privacy, comitati etici, agenzie del farmaco);
- le diverse modalità di archiviazione dei dati presso i promotori degli studi stessi, e, di conseguenza, gli opportuni standard per permettere il dialogo tra sistemi informativi transfrontalieri;
- la necessità di allineare le esigenze dei promotori degli studi, sia pubblici che privati (prevalentemente le industrie farmaceutiche).
LE SOLUZIONI PROPOSTE
Alcuni principi e raccomandazioni sono stati proposti dal gruppo di lavoro del Multi-Regional Clinical Trials Center del Brigham and Women’s Hospital e di Harvard (MRCT Center). Dalle loro ricerche risulta chiaro che una maggiore trasparenza, in generale, e la restituzione dei risultati in particolare, sono aree di interesse in crescita e consentono un maggiore coinvolgimento dei pazienti nel processo decisionale.
La generazione di risultati individuali, che possono essere restituiti ai partecipanti alla ricerca, può avvenire già durante il pre-screening per l’idoneità allo studio ed estendersi oltre la fine della sperimentazione clinica. Ciò include risultati urgenti e risultati incidentali urgenti (tipo A), risultati di laboratorio di routine e risultati incidentali non urgenti (tipo B), risultati individuali di fine studio, compresa l’assegnazione al trattamento o al braccio di studio e gli endpoint primari dello studio (tipo C), risultati esplorativi che possono essere accessori agli endpoint dello studio (tipo D) e risultati aggregati (tipo E). In particolare, esiste un obbligo etico (e medico) impellente di restituire tempestivamente i risultati urgenti e i risultati incidentali urgenti (dati di tipo A). Se opportuno, il protocollo e il documento di consenso informato dovrebbero prevedere queste eventualità e soluzioni operative. Sebbene vi sia una diversità di opinioni sull’obbligo etico di restituire i risultati individuali, il centro MRCT ha raccomandato di differenziare le procedure secondo i seguenti principi:
- I risultati di tipo A (risultati urgenti e risultati incidentali urgenti) devono essere sempre restituiti al medico curante o al partecipante, non appena ne sia confermata la validità, siano essi al di fuori degli intervalli normali e siano associati a una necessità urgente di restituzione a causa delle potenziali conseguenze per la diagnosi, il trattamento o la cura dell’individuo;
- Per i risultati di tipo B (risultati di routine e risultati incidentali non urgenti), il bilancio dei potenziali benefici per il singolo partecipante deve essere soppesato rispetto ai requisiti di risorse e alla fattibilità dell’attuazione della restituzione dei risultati di routine;
- Per il tipo di dati C (risultati individuali di fine studio), come minimo e se possibile, i partecipanti alla ricerca devono ricevere informazioni sull’assegnazione del braccio di studio a cui hanno partecipato dopo la conclusione dello studio. Inoltre, la comunicazione degli endpoint primari deve essere offerta alla fine dello studio, a meno che la restituzione di questi dati non comprometta l’integrità dello studio o degli studi in corso;
- I dati di tipo D (risultati esplorativi) devono essere gestiti caso per caso dallo sperimentatore;
- Per i risultati di tipo E (risultati aggregati), devono essere restituiti in un riepilogo degli endpoint primari e dei dati di sicurezza importanti per i risultati complessivi degli studi, in conformità con le leggi e le linee guida applicabili, ad esempio le linee guida dell’Unione Europea (Health Research Authority ).
La decisione del se, quando e come restituire i singoli risultati si dovrebbe basare almeno in parte sulla natura dei risultati ottenuti e sulla loro importanza. In particolare, i principi chiave e le raccomandazioni non riguardano la restituzione dei risultati aggregati dello studio clinico (Tipo E), bensì sulle altre tipologie.
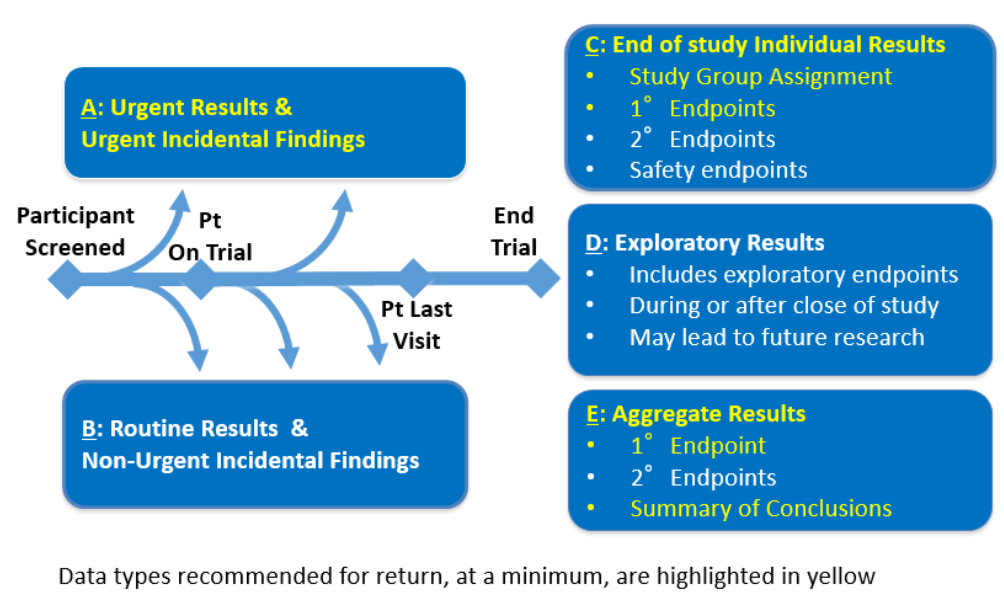
I singoli risultati vengono generati in contesti e in momenti diversi durante la sperimentazione. Ogni volta che un risultato convalidato è perseguibile dal punto di vista medico, vi dovrebbe essere la responsabilità etica di restituire il risultato in questione direttamente al medico o ai medici che hanno la responsabilità primaria della cura dell’individuo o del partecipante, documentando la comunicazione e il trasferimento di responsabilità. Alcune raccomandazioni per i decisori delle autorità regolatorie:
- Fornire i risultati della ricerca individuale risponde agli interessi e alle aspettative espresse da molti partecipanti agli studi clinici, che chiedono che i risultati vengano loro comunicati
- Le considerazioni relative alla restituzione dei risultati della ricerca individuale ai partecipanti alla sperimentazione clinica devono essere integrate nella sperimentazione clinica e pianificate in modo proattivo
- Il processo di consenso informato deve includere informazioni sull’intenzione dello sponsor dello studio clinico in merito alla restituzione dei risultati della ricerca e consentire di discutere le preferenze dei partecipanti in merito alla ricezione di tali risultati
- Il piano per la restituzione dei risultati della ricerca individuale deve essere esaminato da un organismo etico indipendente che supervisiona la ricerca per garantire la tutela dei diritti e del benessere dei partecipanti alla ricerca
- Se i risultati vengono offerti, i partecipanti devono poter scegliere se ricevere o meno i risultati della ricerca individuale
- Gli sponsor e gli sperimentatori hanno l’obbligo di agire in modo responsabile quando restituiscono i risultati individuali, tenendo conto del significato medico, della validità analitica e dell’utilità personale
- I risultati individuali della ricerca devono essere restituiti in modi e tempi che mantengano l’integrità della ricerca, nella misura in cui la sicurezza e il benessere dei partecipanti alla ricerca non sono a rischio
- Lo scopo della ricerca non è l’assistenza clinica e la restituzione dei risultati della ricerca individuale, non può sostituire un’assistenza e una consulenza clinica adeguata
- La restituzione dei singoli risultati della ricerca deve essere pianificata ed eseguita in conformità alle politiche istituzionali, alle leggi e alle normative locali, regionali e nazionali
Questi principi sono “raccomandazioni di base” minime per la restituzione dei risultati individuali della ricerca ai partecipanti allo studio che hanno manifestato la volontà di voler ricevere i risultati. Attualmente si sta lavorando a livello europeo per trovare soluzioni tecnologiche in conformità con il GDPR e il CTR. La recente proposta della Commissione Europa di un Regolamento per la gestione dei dati sanitari potrebbe definire modalità e soluzioni per la restituzione e il riuso dei dati degli studi clinici.
Per approfondimenti: Return of individual results to participants, MRCT Center Harvard; progetto Horizon2020-IMI2-FACILITATE